A Customizable Script for Hybrid Triangles from Genotype Likelihoods
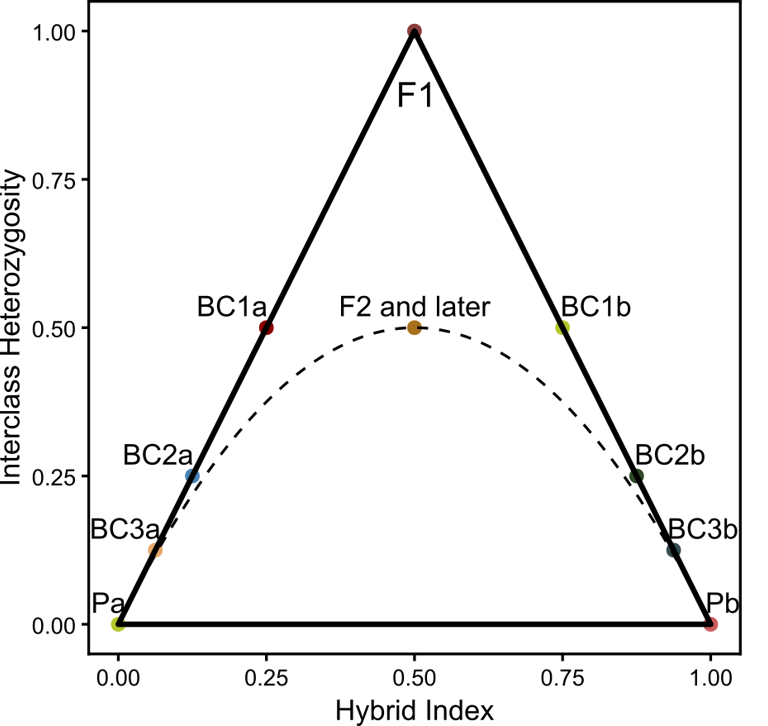
Hybrid triangles are a reliable tool to infer the demographic history of hybrid populations. By incorporating the hybrid index (γ) and the interclass heterozygosity between the parents, this analysis distinguishes F1 from multi-generational hybrids and exposes backcrossing with either parent taxon. Established tools rely on VCF files for hybrid triangle generation, necessitating the hard-calling of SNPs. Here, I present a customizable script to perform the hybrid triangle analysis on genotype likelihood using a multi-step approach in ANGSD.
BIOINFORMATICSADMIXTURE
Tim L. Heller
12/4/20255 min read
How to calculate hybrid triangles from shallow genomes?
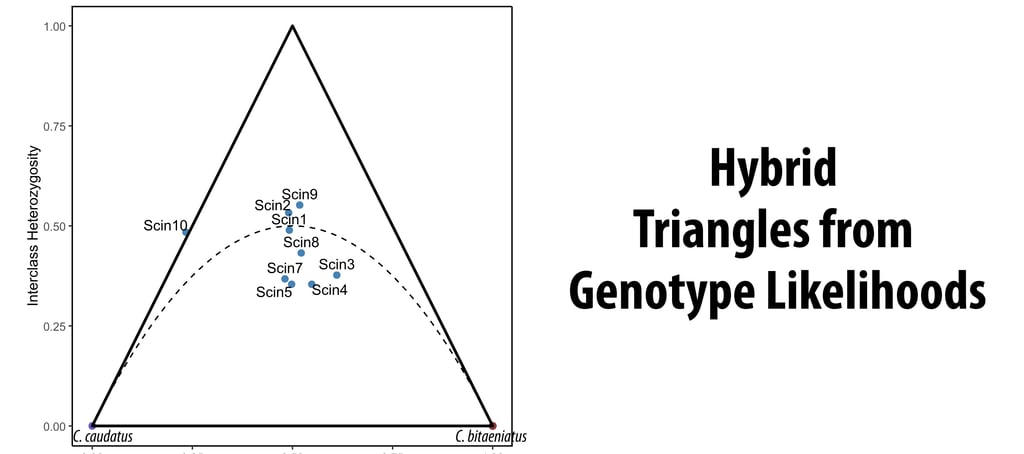
As I was recently confronted with a hybrid dataset of extremely low coverage (2-3X), I developed a customizable script to infer hybrid triangles from genotype likelihoods using ANGSD allele state inference and NGSadmix. In brief, I determined fixed (minor allele frequency > 0.95), divergent sites relative to an ancestral reference sequence for both parental taxa and performed individual SNP calling on this subset of sites for the hybrid swarm. To calculate the hybrid index (γ), I ran NGSadmix on the hybrid parents and the hybrids with K = 2 and extracted individual admixture proportions in each hybrid individual.
But why go through all that trouble? First, hybrid triangles might be the most straightforward way to establish, if a hybrid has backcrossed either to itself or with either one of the parents. This is particularly useful to determine, if a hybrid population is fertile (non-fertile hybrids would not reproduce past F1) and if there is still genetic exchange with the parents. In F1 hybrids, we expect equal contribution from both parents (γ = 0.5), as recombination between the genetic material of the parents had no time to act. Additionally, in diploid organisms, we assume all divergent fixed sites between the parents to be heterozygous in the hybrid's genome. This immediately exposes F1 hybrids in the triangle. Further backcrossing to itself might lead to deviations in γ and according to the Hardy-Weinberg equilibrium to an interclass heterozygosity of 0.5 (p = 0.5, q = 0.5; i.e., heterozygosity = 2pq). If a hybrid backcrosses to either parent, γ will be shifted towards that direction.
To start calculating hybrid triangles, we can first run NGSadmix on our hybrids together with both parents. It is advised to keep sample sizes of all three groups in a similar magnitude to avoid biases in ADMIXTURE analysis.
NGSadmix requires beagle-type likelihoods which can be generated from .bam-files with ANGSD like:
angsd -nThreads 6 -bam ./path/to/list/of/bam/files -out ./out/prefix -rf ./path/to/list/of/regions/chromosomes -uniqueOnly 1 -minMapQ 20-minQ 20 -GL 2 -doGlf 2 -doMajorMinor 1 -skipTriallelic 1 -doMaf 1 -minMaf 0.05 -SNP_pval 1e-6 -doCounts 1 -setMinDepthInd 1
Then, we execute NGSadmix.
./NGSadmix -likes /path/to/beagle/gz -K 2 -o ./out_prefix -P $threads
We will keep this output-file (.qopt) for later. However, you need to make sure to keep the order of individuals as provided in the bam-list (-bam) for the beagle likelihoods.
The next step is a bit more complex. We need to run multiple scripts to get this to work properly. We first repeat the complete SNP calling on both parents by setting an outgroup as a reference to determine the divergent sites between the hybrid parents. By setting an outgroup, we ensure that ANGSD does not skip the fixed (monomorphic) sites in either parent population. Of course, many of these sites have diverged from the outgroup in the most recent common ancestor of the parent species which is why we join this list afterwards. All that we need is a list of the sites that are different (diverged) between the parent.
conda activate angsd
## Infer diverged, fixed sites in both parents
files=("Europe" "JuandeNova")
for file in "${files[@]}"; do
echo "Processing file: ${file}_BAM.list"
angsd \
-nThreads 4 \
-bam "${file}_BAM.list" \
-out /dss/dsslegfs01/pr53da/pr53da-dss-0039/crypto/03.variant_calling/code/00.angsd/Fst/Heterozygosity/Tromelin/"${file}" \
-GL 2 -doMaf 1 -minMapQ 20 -doMajorMinor 5 -doCounts 1 -minQ 20 -remove_bads 1 -minMAF 0.95 -SNP_pval 1e-3 -minInd 10 \
-anc /dss/dsslegfs01/pr53da/pr53da-dss-0039/crypto/02.reads_mapped/code/nf-umap/reference/rCryege1.pri.20221122.fasta
done
cd Heterozygosity/Tromelin/
zcat Europe.mafs.gz > Europe.mafs
zcat JuandeNova.mafs.gz > JuandeNova.mafs
## Input .mafs files
EUROPE="Europe.mafs"
JUANDE="JuandeNova.mafs"
## Output file
OUTPUT="fixed_diff_sites.txt"
## Extract chromo, position, minor allele from each .mafs file
awk 'NR > 1 {print $1"\t"$2"\t"$4}' "$EUROPE" > europe.minor
awk 'NR > 1 {print $1"\t"$2"\t"$4}' "$JUANDE" > juande.minor
### Find sites fixed for different alleles
awk '
FNR==NR {
allele[$1 FS $2] = $3
next
}
{
key = $1 FS $2
if (key in allele && allele[key] != $3)
print $1"\t"$2}
' europe.minor juande.minor | sort -V > "$OUTPUT"
## Clean up intermediate files (optional)
rm europe.minor juande.minor
echo "Done. Divergent fixed sites written to $OUTPUT"
angsd sites index fixed_diff_sites.txt
Subsequently, we employ this list to calculate the interclass heterozygosity in the hybrids with the following script:
conda activate angsd
angsd sites index fixed_diff_sites.txt
## Set paths
BAM_LIST="../../Tromelin_no6_BAM.list"
SITES_FILE="fixed_diff_sites.txt"
ANGSD_PATH="angsd" # If ANGSD is in your PATH, otherwise provide full path
INTERCLASS_OUTPUT="interclass_heterozygosity.txt"
rm $INTERCLASS_OUTPUT
for files in ./*.mafs.gz; do
name=$(basename $files .mafs.gz)
sites=$(zcat $files | wc -l)
Hets=$(zcat $files | awk '$5 > 0.45 && $5 < 0.55' | wc -l)
if [ "$sites" -ne 0 ]; then
ratio=$(echo "scale=5; $Hets / $sites" | bc)
else
ratio="NA"
fi
echo -e "$name\t$Hets\t$sites\t$ratio" >> $INTERCLASS_OUTPUT
done
Now, with the list of the samples for the NGSadmix run (check -bam list -> Popfile.txt), the admixture proportions in the .qopt file (hybrids.qopt) from NGSadmix, and the interclass_heterozygosity.txt, we can switch over from Linux to R.
library(ggplot2)
library(tidyr)
libary(dplyr)
setwd("~/Thesis/Hybrid Triangle")
interclass = read.table("Interclass_Heterozygosity.txt", sep = "\t")
admix = read.table("hybrids.qopt", sep = "\t")
Pops = read.table("Popfile.txt", sep="\t")
final = cbind(Pops,admix)
colnames(final) = c("Sample", "Pop", "V1","V2")
palette_expanded_3 <- c(
"#8B0000", # Dark Charcoal
"#B4C72B", # Dark Olive Green (Replaced Dark Salmon)
"#4682B4", # Steel Blue
"#2E4A25", # Dark Forest Green
"#E7A86B", # Terra Cotta
"#2F4F4F", # Dark Slate Gray
"#8B3E3E", # Mahogany
"#A76E1B", # Saddle Brown
"#B4C72B", # Olive Drab
"#CD5C5C", # Indian Red
"#8B0000", # Dark Red
"#708090", # Slate Gray
"#6A5ACD", # Slate Blue
"#C0C0C0" # Silver
)
Tromelin = final %>% filter(Pop=="TR") %>% cbind(interclass)
colnames(Tromelin)=c("Sample", "Pop", "V1", "V2", "Sam", "Het", "Sites")
Tromelin$ICH = Tromelin$Het / Tromelin$Sites
Rest = final %>% filter(!Pop=="TR") %>% mutate(ICH = 0)
Tromelin_filt = Tromelin[,c(1,2,3,4,8)]
Final = rbind(Rest, Tromelin_filt)
# Open triangle (without bottom edge)
triangle_df <- data.frame(
x = c(0.5, 1, 0),
y = c(1, 0, 0)
)
# Hardy-Weinberg Equilibrium (HWE) curve: Interclass heterozygosity (p12) as a function of hybrid index (p1)
p1_vals <- seq(0, 1, length.out = 100) # Hybrid index values from 0 to 1
hwe_df <- data.frame(
x = p1_vals,
y = 2 p1_vals (1 - p1_vals) # HWE formula for interclass heterozygosity (p12)
)
# Plot
ggplot() +
geom_point(data = Final, aes(x = V1, y = ICH, color = Pop), size = 2) +
geom_polygon(data = triangle_df, aes(x = x, y = y), fill = NA, color = "black", linewidth=1) + # Open triangle
geom_line(data = hwe_df, aes(x = x, y = y), linetype = "dashed", color = "black") + # HWE curve
geom_text(data = filter(Final, Pop == "C. sp. \"Tromelin\""), aes(x = V1, y = ICH, label = Sample), vjust = -0.5, color = "black") + # Label TR samples
coord_fixed(ratio = 1) +
labs(x = "Hybrid Index", y = "Interclass Heterozygosity") +
theme_classic() + scale_color_manual(values=palette_expanded_3)
Please let me know, if it has worked for you too. I am happy to help if problems arise!
How to cite this article?
Heller, T. L. (2025). A Customizable Script for Hybrid Triangles from Genotype Likelihoods. Tim L. Heller. https://www.timlheller.com/hybrid-triangles